Biosim lab tips
Generate ssh key for UB CCR
To use the secure shell (SSH) to connect to CCR’s login servers, you will need to generate a new SSH key on your local machine:
ssh-keygen -t ed25519 -C "email@buffalo.edu"
The -t option specifies which type of secure key to generate. ed25519 refers to speicifc elliptic curve algorithm that you can read more about here. The -C option expects you to add a comment to the key, here you should put your Buffalo email address.
Here is an example:
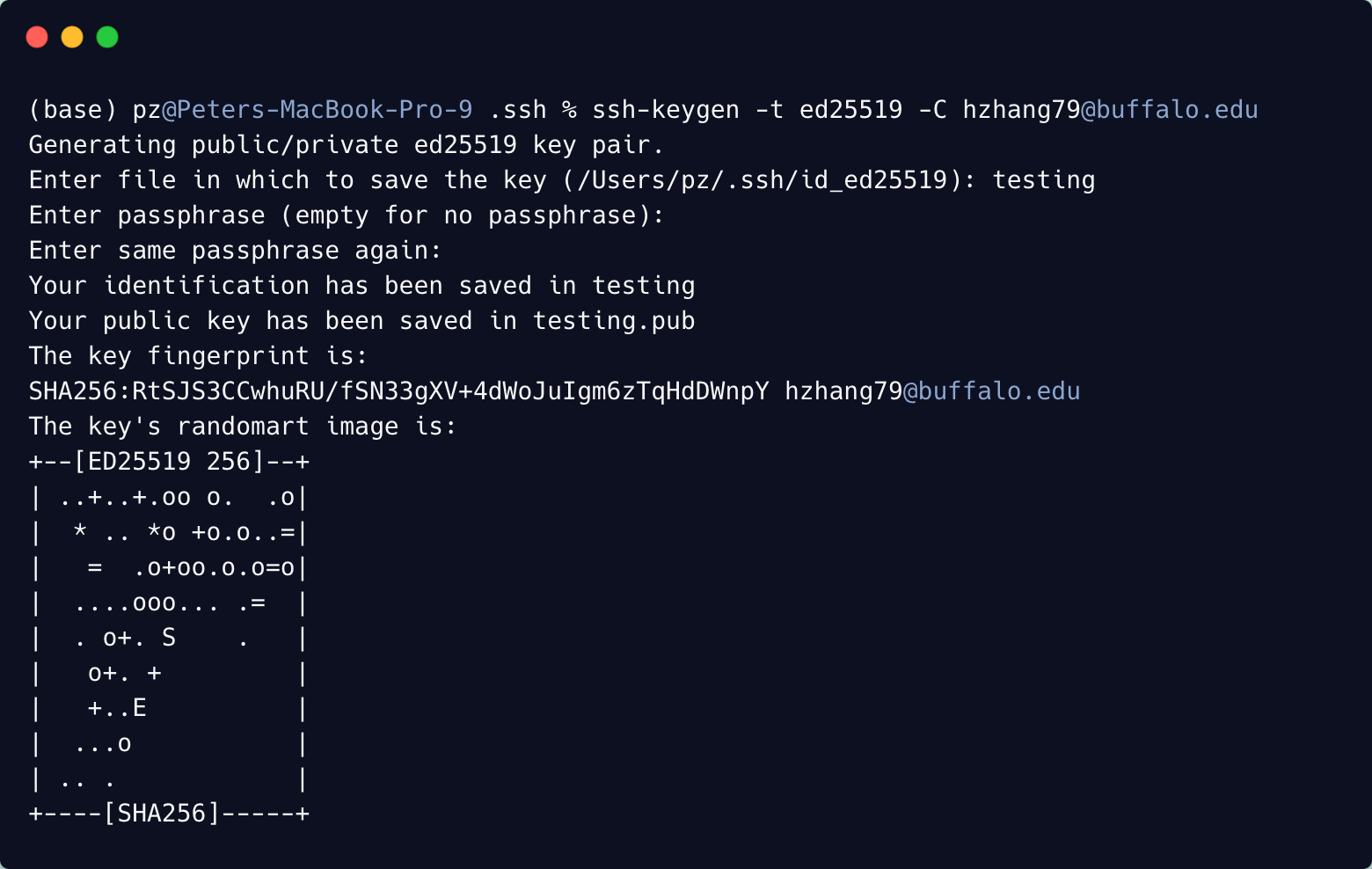
At the end, in the ~/.ssh directory, you should find the public key and the private key. In my case, I have testing.pub as my public key and testing as my private key Copy the content of the public key to the CCR. Wait for approval and then you can login to CCR using ssh. You should do this process for every machine that you wish to connect to CCR from.
Here is an example of my CCR account, I have three machine registered:
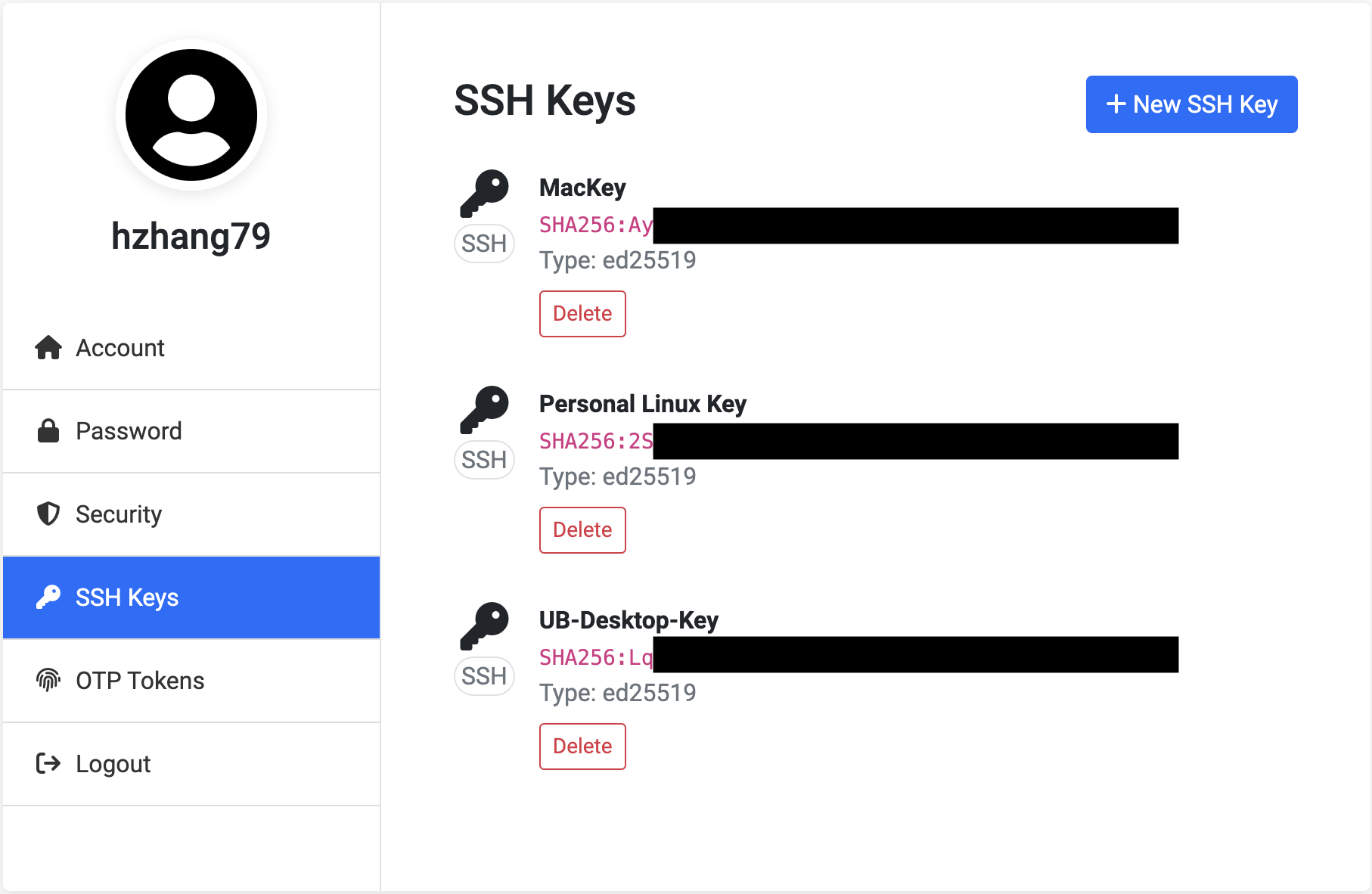
References: Generate New SSH Key
Login to UB CCR and nevigate to our group folder
Now you have uploaded your public key to CCR and your matching private key in the .ssh folder, you can fire up a terminal window and log into CCR with the command below!
ssh -i your-key-name your-username@vortex-future.ccr.buffalo.edu -o ServerAliveInterval=60
The -o ServerAliveInterval=60 option sends a keepalive message to the server every 60 seconds, prevent you from disconnected when idel.
I recommend to add the ssh command to your bash resource file .bashrc or .zshrc if you are on Mac. Nevigate to the file by typing: vim ~/.bashrc, add alias sshccr=ssh -i your-key-name your-username@vortex-future.ccr.buffalo.edu -o ServerAliveInterval=60', then activate it by typing source ~/.bashrc or restart the terminal. Now you can log into CCR by typing sshccr instead of the whole command. Be sure to not delete or change anything else in your bash resource file.
To get to our group project directory:
cd /projects/academic/nguyenh
To get to our scratch directory:
cd /vscratch/grp-nguyenh
Trajectory Alignment using MD Analysis
Sometimes, you might need to align a trajectory to a reference frame and save it for analysis. This can come in handy when calculating things like root-mean-sqaure deviation (RMSD) or root-mean-sqaure fluctuation (RMSF). Here, I’ve shared a script that aligns every frame of an RNA trajectory to its center of mass to make these analyses easier.
import os
import numpy as np
import MDAnalysis as mda
from MDAnalysis import transformations as trans
from MDAnalysis.analysis import align
def align_COM(directory, pdb, dcd)
os.chdir(directory)
# Create an Universe with the trajectory(dcd) and topology file(pdb)
u = mda.Universe(pdb, dcd)
# Define your box size (here all edge is 720 angstrom with 90 degree angles)
dim = np.array([720, 720, 720, 90, 90, 90])
# Select the molecule you want to align (I am working with RNA, so Adenine in my case, you can print out your selection to see if you are selecting your intended molecule)
ADE = u.select_atoms('resname ADE')
# Define your workflow, here I set my box dimension, unwrap all atoms, center ADE's (I defined earlier) center of mass to the center of box, then I wrap all the atoms.
workflow = [trans.boxdimensions.set_dimensions(dim),
trans.unwrap(u.atoms),
trans.center_in_box(ADE, center='mass'),
trans.wrap(u.atoms)]
# use add_transformations to apply the workflow you defined earlier to the trajectory
u.trajectory.add_transformations(*workflow)
# now your trajectory is aligned, to save it I use the align.AlignTraj function. Here I save my new trajectory as "md-align-wrap.dcd"
align.AlignTraj(u, u, select='resname ADE', filename="md-align-wrap.dcd", match_atoms=True).run()
References: Aligning a trajectory to a reference, On-the-fly transformations, Centering a trajectory in the box
Alignment to principal axes
For flexible molecules, it can be useful to align the molecule to its principal axes. I have used this technique to visualize the ion distribution surrounding the flexible single stranded RNA. I do this by utilizing the orient package in VMD, and save the principally aligned trajectory for analysis.
-
Download: orient and la. You will need la, the linear algebra package to run orient.
-
unpack the package:
gunzip orient.tar.gz
tar -xf orient.tar
gunzip la101psx.tar.gz
tar -xf la101psx.tar
- Open VMD and go to VMD TkConsole:
Go to la1.0 folder you got from previous step
cd la1.0
source la.tcl
Go to orient folder you got from previous step
cd orient
source orient.tcl
- I provide the script I have to align the principal axes of my RNA to the z-axis of the box
namespace import Orient::orient
# Get the number of frames in the trajectory
set num_frames [molinfo top get numframes]
# Select all atoms
set sys [atomselect top "all"]
# Set the molecule you are working with, here I select everything that is not Magnesium
set rA [atomselect top "all not name Mg"]
# Iterate over each frame in the trajectory
for {set i 0} {$i < $num_frames} {incr i} {
# Set the frame
animate goto $i
# Compute the principal axes and align the molecule
set I [draw principalaxes $rA]
# I align the first principal axis to the z axis
set A [orient $rA [lindex $I 2] {0 0 1}]
$sys move $A
# I align the second principal axis to the y axis
set I [draw principalaxes $rA]
set A [orient $rA [lindex $I 1] {0 1 0}]
$sys move $A
# Update the coordinates for the current frame
$sys update
$rA update
}
# Write the modified trajectory to a new file
animate write dcd principal-aligned.dcd
References: Alignment to principal axes in VMD
Tips for using Arena to map coarse-grained structures to atomistic structures
The coarse-grained structures can be backmapped to the corresponding atomistic structures with Arena. There are many ways you can use this program to fit your needs, here I provide some of my scripts and procedures for you to use as a guidance.
I extracted 10,000 random-frames from my coarse-grained simulations with the script below - my goal was to eventually calculate 10,000 SAXS profiles to get the ensemble average.
# writen by Peter Zhang - 09/18/2024
# Load the required packages
package require pbctools
package require topotools
package require Tcl
# Define trajectory and output directory
set traj "/home/pz/remote/RNA-homopolymers/rA30/experiment-comparison/Na20/Mg2/rA30-4-1/md-align-wrap.dcd"
set output_dir "/home/pz/Projects/SAXS-calculation-homopolymer/rA30-2mM-calculation/cg-10000-frames"
# Load the trajectory
mol new /home/pz/remote/RNA-homopolymers/rA30/experiment-comparison/Na20/Mg2/rA30-4-1/rA30-Mg2-Na20-1.pdb type pdb
mol addfile $traj type dcd waitfor all first 3000 last -1
# Get the total number of frames in the traj
set num_frames [molinfo top get numframes]
# Set the number of random frames to select
set num_random_frames 10000
# Initialize a list to store the selected frames
set selected_frames {}
# Ensure that the number of requested frames does not exceed the total number of frames
if {$num_random_frames > $num_frames} {
set num_random_frames $num_frames
}
# Randomly select frames without replacement
for {set i 0} {$i < $num_random_frames} {incr i} {
set frame [expr {int(rand() * $num_frames)}]
# Ensure no duplicate frames are selected
while {[lsearch -exact $selected_frames $frame] != -1} {
set frame [expr {int(rand() * $num_frames)}]
}
lappend selected_frames $frame
}
# Export each selected frame as a PDB file with a unique name
foreach frame $selected_frames {
# Set the current frame
molinfo top set frame $frame
set sel [atomselect top "all not name Mg"]
# Define the output file name
set output_file [format "%s/frame_%05d.pdb" $output_dir $frame]
# Write the selected atoms to the PDB file
$sel writepdb $output_file
# Delete the atom selection to free up memory
$sel delete
}
puts "Completed exporting to $output_dir"
Arena will only recognize standard RNA atom naming system (P, OP1, OP2, O5’, C5’, C4’, O4’, C3’, O3’, C2’, O2’, C1’, N9, C8, N7, C5, C6, N6, N1, C2, N3, C4) - so if you named your coarse-grained beads differently, you need to change the bead name. For my three bead system, I chose to map the phosphate bead to the phosphate atom (P), the sugar bead to C2’, the nucleobase to C4. If you also named your residue differently in the coarse-grained pdb file, you should change them to the one letter code (A, U, C, or G).
The naming change can be done using the stream editor (sed) from your terminal, below is an example:
sed -i 's/\(P___\)ADE/\|_____A/' *.pdb
If after those changes, Arena cannot recognize your naming, make sure to check the spacing.
Now, once you input those coarse-grained structures and have the output from Arena. You can load them to vmd to visualize them. And you will likely see “bad bonds”, they are either too long or they are merged with some other atoms. To correct those “bad bonds”, we will need to perform an energy minimization step. I use GROMACS and Amber force field χOL3. Here is the script:
#!/bin/bash
# Written by Peter Zhang
# 07/23/24
# Referenced from Magdalena A. Jonikas (RNA gromacs tips)
FILE_PATTERN=$1 # pattern to match pdb files
NP=$2 # number of ions to add
export GMX_MAXBACKUP=-1
for FILENAME in $FILE_PATTERN; do
NAME="${FILENAME%.pdb}"
if [ ! -e $NAME-em-final.pdb ]; then
echo "Running $NAME"
echo "Checking for needed files"
if [ ! -e em.mdp ]; then
echo "Missing em.mdp"
exit 1
fi
if [ ! -e $NAME.pdb ]; then
echo "Missing $NAME.pdb"
exit 1
fi
echo "Fixing the pdb file syntax"
if head -n 3 $NAME.pdb | grep -E '(P|OP1|OP2)' > /dev/null; then
echo "Deleting the first three lines of $NAME.pdb"
sed -i '1,3d' $NAME.pdb
fi
echo "Making topology file from pdb file"
gmx pdb2gmx -f $NAME.pdb -p $NAME.top -o $NAME.gro -ff amber03 -water tip3p -ignh >& out-pdb2gmx-$NAME.txt
if [ ! -e $NAME.gro ]; then
echo "FAIL: pdb2gmx"
exit 1
fi
echo "Making box and adding water"
gmx editconf -f $NAME.gro -o -d 2.0
gmx solvate -cp out.gro -cs spc216.gro -p $NAME.top -o $NAME-solv.gro >& genbox-solvate-$NAME.txt
if [ ! -e $NAME-solv.gro ]; then
echo "FAIL: editconf or solvate"
exit 1
fi
echo "Performing first energy minimization"
gmx grompp -v -f em.mdp -c $NAME-solv.gro -o $NAME-em -p $NAME.top -maxwarn 10 >& grompp-em-$NAME.txt
if [ ! -e $NAME-em.tpr ]; then
echo "FAIL: grompp em"
exit 1
fi
gmx mdrun -v -s $NAME-em -o $NAME-em -g $NAME-emlog >& out-mdrun-em-$NAME.txt
if [ ! -e $NAME-em.trr ]; then
echo "FAIL: mdrun em"
exit 1
fi
# Add ions (30 for homopolymers with 30 nucleotides)
echo "Adding ions"
echo 3 | gmx genion -s $NAME-em.tpr -o $NAME-ion -p $NAME.top -np $NP >& out-genion-$NAME.txt
if [ ! -e $NAME-ion.gro ]; then
echo "FAIL: genion"
exit 1
fi
echo "Performing final energy minimization"
gmx grompp -v -f em.mdp -o $NAME-em-final -c $NAME-ion.gro -p $NAME.top -maxwarn 10
if [ ! -e $NAME-em-final.tpr ]; then
echo "FAIL: grompp em-final"
exit 1
fi
gmx mdrun -v -s $NAME-em-final -o $NAME-em-final
if [ ! -e $NAME-em-final.trr ]; then
echo "FAIL: mdrun em-final"
exit 1
fi
echo "Converting trajectory to pdb"
echo 1 | gmx trjconv -f $NAME-em-final.trr -s $NAME-em-final.tpr -o $NAME-em-final.pdb >& out-convert-$NAME.txt
if [ ! -e $NAME-em-final.pdb ]; then
echo "FAIL: em convert"
exit 1
fi
echo "Completed processing $NAME"
fi
done
For additional details, please check out these references:
Remotely access lab desktop and NAS
Prerequisite: If you are off-campus, you must connect to UB VPN before proceeding.
Accessing the NAS from the Lab Desktop
From your lab workstation, you can directly access the NAS by running:
cd ~/mnt/nas/projects
If this is your first time accessing the NAS, please create a folder with your name:
mkdir ~/mnt/nas/projects/<your_name>
Remotely Access Lab Desktop
To remotely connect to your lab computer, first determine its IP address by running:
hostname -I
You should see output similar to:
(base) hzhang79@cast-nguyenh-wk1:~$ hostname -I
xxx.xxx.xxx.xx xxx.xxx.xxx.xxx xxxx:cc:xxxx:xxx::xxxx
Explaination of the IP Address:
xxx.xxx.xxx.xxUniversity at Buffalo network IP (used for remote access).xxx.xxx.xxx.xxxPrivate/local network IP (only works within the same local network).xxxx:cc:xxxx:xxx::xxxxIPv6 address Note: Portions of the IP addresses are masked (x) for privacy.
Connecting to Your Lab Desktop via SSH
From your personal computer, use the UB network IP (xxx.xxx.xxx.xx) to connect:
ssh username@xxx.xxx.xxx.xx
For example:
ssh hzhang79@xxx.xxx.xxx.xx
Enabling SSH Access (if connection fails)
If the connection fails, SSH access may not yet be enabled on your workstation. In that case, send a request to:
cas-support@rt.caset.buffalo.edu
Email Template
Dear Chris, I am trying to set up remote access to my lab workstation (cast-nguyenh-xxx). Replace this with your computer identifier (found on the white sticker on the machine). IP address: xxx.xxx.xxx.xx Could you please enable SSH access (openssh-server) and allow port 22? Thank you! Your Name
Once SSH access (port 22) is enabled, you can connect to your lab desktop remotely just as if you were physically sitting in front of it (via command line).